pip install alphapeptstats or download it from GitHub.
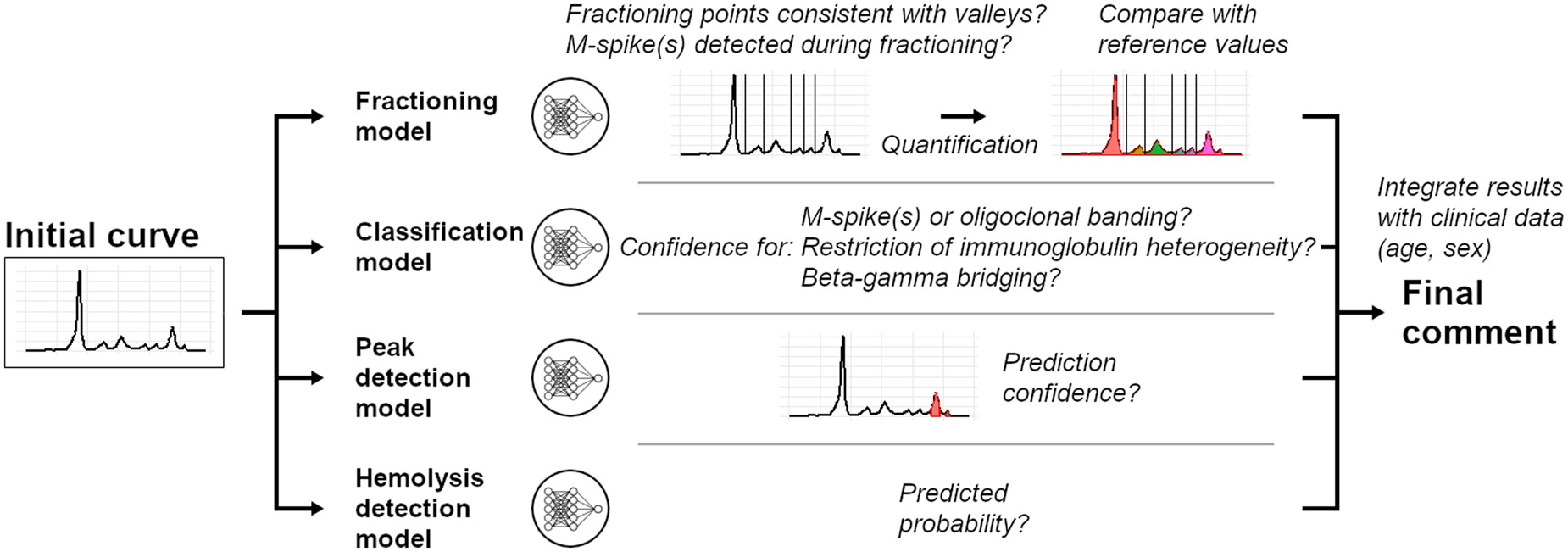
Serum protein electrophoresis (SPE) is a critical diagnostic test used primarily for detecting and monitoring monoclonal gammopathies. Traditional SPE interpretation can be time-intensive and is often subject to variability due to human expertise. SPECTR, developed by Floris Chabrun and collaborators, is an AI-driven tool that automates and standardizes SPE interpretation by generating clinically relevant text comments directly from raw SPE data. This deep learning model is trained to achieve expert-level accuracy, improving the consistency and reliability of SPE analyses.
About SPECTR
SPECTR (Serum Protein Electrophoresis Computer-Assisted Recognition) uses a deep learning model to analyze SPE curves, identifying both quantitative and qualitative abnormalities. Built on an extensive, expert-annotated dataset of nearly 160,000 SPE entries, SPECTR can interpret key diagnostic indicators such as M-spikes, immunoglobulin heterogeneity, and beta-gamma bridging. Validated on an independent cohort of 70,000 SPEs, SPECTR demonstrates remarkable accuracy and consistency, making it a powerful tool for high-throughput clinical applications.
Key Features
- AI-Powered Interpretation: Uses deep learning to generate expert-level interpretations directly from SPE curves.
- High Accuracy: Achieves a ROC-AUC ≥ 0.99 in detecting abnormalities, with inter-rater reliability surpassing human expert agreement.
- Open Access and Online: Freely accessible via an online application, allowing real-time, editable validation assistance for clinical use.
- Transparent Architecture: Designed to avoid the “black box” effect, following academic guidelines for SPE interpretation.
Design and Components
SPECTR’s design leverages AI to enhance the accuracy and reproducibility of SPE interpretation:
- Deep Learning Models: SPECTR relies on pre-trained models that were developed using over 150,000 SPE entries, enabling it to detect subtle variations in SPE patterns.
- Data Processing and User Interface: The application is built with R (app.R script) for the user interface and Python (ai.py script) for model inference. This design provides a user-friendly interface while allowing robust backend processing.
- Training Resources: The repository includes scripts and example datasets used for training, supporting further model refinement or adaptation for specific lab requirements.
Performance and Results
In validation tests, SPECTR demonstrated high agreement with human experts, achieving a kappa value of 0.632, which is higher than inter-expert agreement (κ = 0.624). Its accuracy in detecting M-spikes and other abnormalities reaches a ROC-AUC of ≥ 0.99, showcasing its potential as a reliable alternative to manual interpretation. SPECTR’s consistency and resilience to minor variations make it ideal for high-throughput clinical use.
How to Use SPECTR
- Access the Online Application: Use SPECTR directly online at https://spectr.shinyapps.io/SPECTR.
- Upload SPE Data: Input raw SPE curve data for analysis.
- Receive AI-Generated Interpretation: SPECTR generates interpretable comments on SPE patterns, assisting in clinical decision-making.
- Edit as Needed: Practitioners can review and modify the interpretations to ensure diagnostic relevance.
For a deeper dive into SPECTR’s design, users can access the full original article in Clinical Chemistry via this link.
SPECTR represents a significant advancement in the standardization of SPE interpretation, offering a high-accuracy, AI-powered solution to streamline the analysis of complex protein patterns. By automating the interpretation process, SPECTR supports clinical labs in achieving reliable, reproducible results, ultimately contributing to better patient care. Its open-access nature further allows continuous improvement and adaptation by the clinical community.
